plastic deformation을 보기 위해 사용하는 다양한 simulation model들이 있다.
다양한 length scale과 time scale에 따라 사용하는 model이 달라진다.
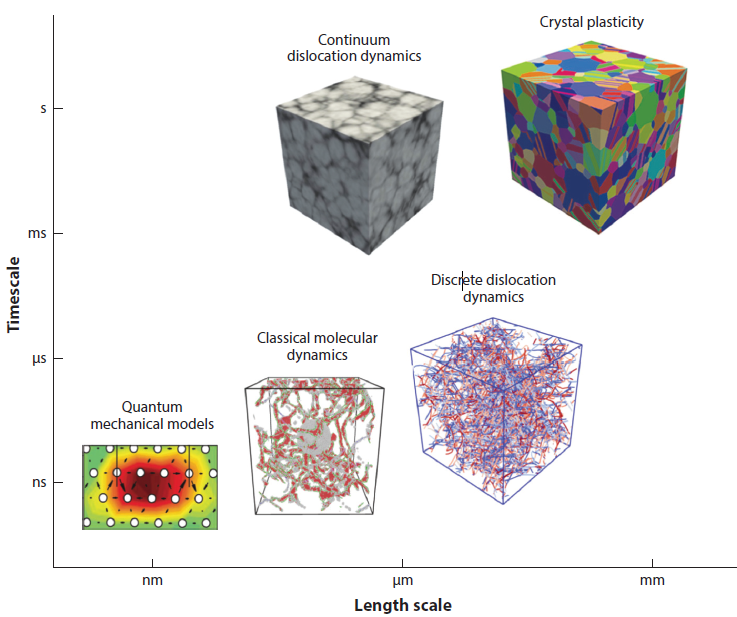
그런데, 왜 scale에 따라 사용하는 model이 달라지는 것일까?
예를 들어 설명해 보겠다.

plastic deformation이 일어나 dislocation network(서로 상호작용하는 dislocation들의 집합체)이 생기면, cellular structure 가 생긴다.(cellular structure가 생기는 이유도 차후에 설명해 보겠다.) 이러한 cell의 평균적인 크기는 1 μm 의 order를 가지고 있다.[2]
이 정도 scale의 bulk plastic deformation을 이해하기 위해 시뮬레이션을 돌린다고 생각해보자.
만약, atomistic simulation을 사용한다고 하면, 이 정도 scale에서 1010개의 atom들을 다루어야한다..!! 즉 계산량이 너무 많고, 시간도 많이 걸릴 것이다.
*참고
Although the computational cost of classical MD scales linearly with the number of atoms, the cost of simulating large atomistic systems is still high. Today, MD simulations on dislocations are usually performed for systems containing up to ∼106 atoms using ∼100 central processing units (CPUs). Given that MD time steps are typically on the order of 1 fs, the time duration of MD simulations is on the order of ∼10 ns (assuming 107 steps), leading to very high strain rates (∼107 s−1, assuming 10% strain over 10 ns). In order to overcome the timescale limitation, a number of advanced techniques for computing transition/activation rates have been applied to thermally activated dislocation processes such as nucleation and obstacle bypass. Furthermore, accelerated MD methods have shown promising results in the dynamics of point defects in crystals, but their application to dislocations (being extended defects) has been dificult. [1]
- 최근에는 Atomistic simulation에서도 timescale limitation을 극복하기 위한 발전이 이루어 지고 있긴 하다..!
따라서 이러한 bulk size의 plastic deformation을 보기 위해서는 atomistic simulation은 현실적으로 불가능 한 것 이다.
우리는 atomistic approch를 통한 분석이 아닌 다른 모델링을 통한 분석의 필요성을 느끼게 되었다.
Mesoscale에서의 새로운 모델링인 Dislocation Dynamics이 필요하다!!
Dislocation Dynamics 에서는 atomistic approach와 뭐가 다르길래 좀 더 큰 scale에서 분석이 가능 한 것일까?
atomistic approach에서는 모든 atom들을 고려하지만, DD approach에서는 dislocation line의 interaction과 motion이 주요한 factor로 작용하기 대문에, 계산량이 atomistic approach에 비해 대폭 줄어들게 된다. 10 μm의 length scale과 1 ms 의 time scale을 가지게 된다.
궁극적으로는, lower scale(atomistic approach; Molecular dynamics)에서의 key feature를 찾아 higher scale(DD, Crystal plasticity;CP)의 인풋 데이터로 활용하여, 좀 더 본질적인 분석을 완성시키는 것이 목표이다.
DD는 실험적으로나 fundamental한 model들을 통해 얻은 dislocation line의 behavior를 필요로 하는 데, 다음 포스트를 통해 알아보겠다.
[1] Bulatov, V. V., & Cai, W. (2006). Frontiers in the simulation of dislocations. Annual Review of Materials Research, 36, 1-38. https://doi.org/10.1146/annurev.matsci.36.090804.094515
[2] Y. Kawasaki, T. Takeuchi, Cell structures in copper single crystals deformed in the [001] and [111] axes. Scr. Met. 14, 183-188 (1980)
[3] Sills, R. B., Kuykendall, W. P., Aghaei, A., & Cai, W. (2020). Chapter 2: Fundamentals of dislocation dynamics simulations. In Dislocation Dynamics Handbook (pp. 35–65). Springer.
공부, 기록에 초점을 둔 글입니다.
틀린 내용이나 다른 문제가 있다면 댓글에 남겨 주세요. 많은 도움이 될 것 같습니다!